Chapter 2 Introduction
The microeco package has several advantages compared to other packages in R.
The primary objective behind the development of this package is to assist users in rapidly analyzing microbiome data and
utilizing a range of cutting-edge and commonly adopted methodologies.
To facilitate data mining, every part of the microeco package has been modularized to ensure that users can easily recall, search, and employ classes.
It is important to note that, in addition to being demonstrated in the tutorial,
users can also save intermediate data within each object and utilize them with other tools according to formatting necessities.
Main data stored in the object of each class are the frequently-used data.frame format,
thereby making it effortless to save, modify, and employ intermediate and outcome files with other tools.
Prior to exploring the specific usage of each class, we first introduce a few critical points.
2.1 Framework
This is a rough framework for users to fast understand the design of microeco package.
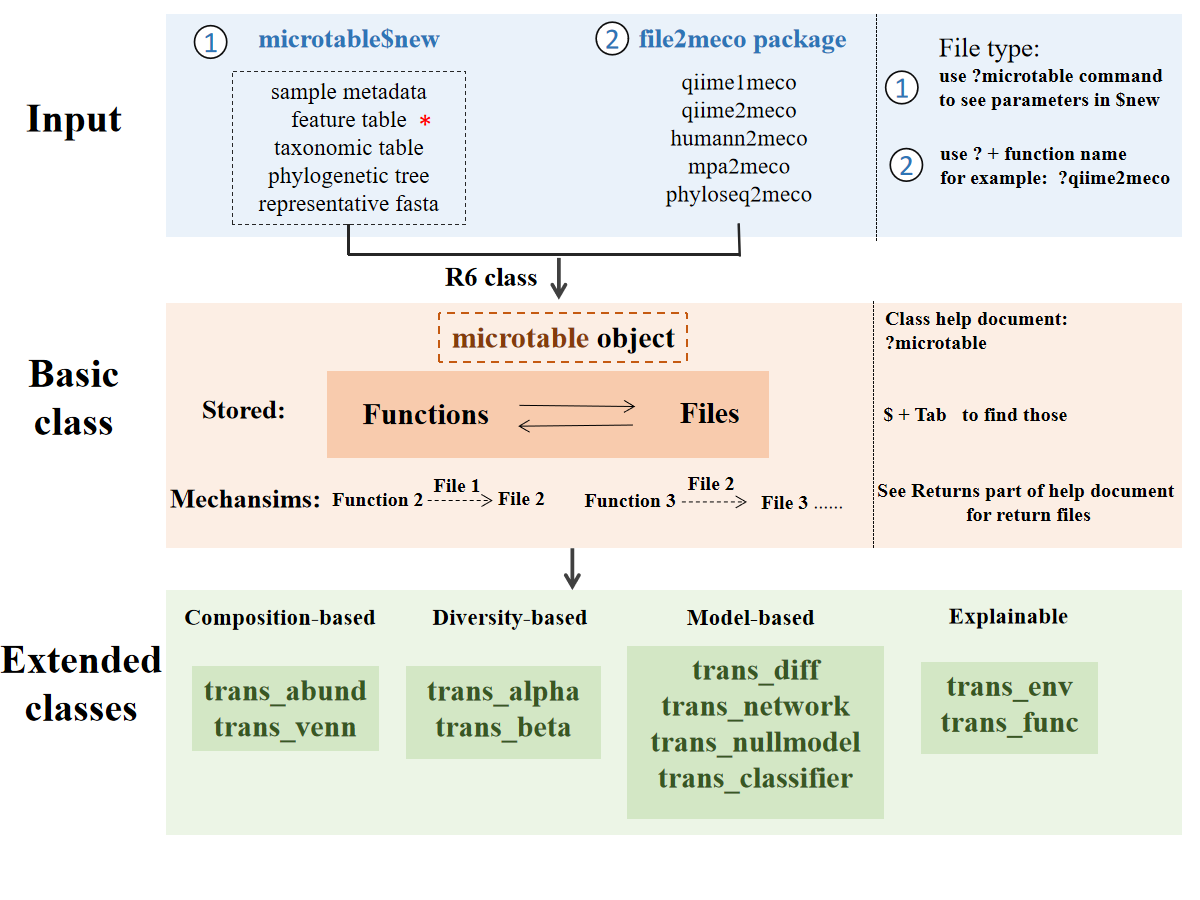
The stored ‘Functions’ and ‘Data’ represent that the user can access those functions or data in R6 object using $ operator
as shown in the figure. An example is the function dataset$cal_alphadiv() and its return result dataset$alpha_diversity.
The dataset is a microtable object.
The return of functions is generally named with the prefix ‘res_’ to make users easily find them when using Rstudio and the keyboard shortcuts (Tab).
Except for microtable class, the transformed data in created object is generally named with the prefix ‘data_’.
2.2 R6 Class
All the main classes in microeco package depend on the R6 class (Chang 2020). R6 uses the encapsulated object-oriented (OO) programming paradigm, which means that R6 is a profoundly different OO system from S3 and S4 because it is built on encapsulated objects, rather than generic functions. If the user is interested in the class features, read more from ‘Advanced R’ book (https://adv-r.hadley.nz/).
A generic is a regular function, so it lives in the global namespace. An R6 method belongs to an object so it lives in a local namespace. This influences how we think about naming. The methods belong to objects, not generics, and the user can call them like object$method().
R6’s reference semantics allow methods to simultaneously return a value and modify an object.
Every R6 object has an S3 class that reflects its hierarchy of R6 class.
2.3 Help
The usage of help documents in the microeco package may be a little different from other packages we often used. If the user wish to see the help document of a function, please search the name of the class it belongs to (not the name of the function) and click the link of the function.
# first install microeco, see https://github.com/ChiLiubio/microeco
# load package microeco
library(microeco)# show all the classes and tutorial links
?microeco
# show the detailed description of the class microtable
# same with: help(microtable)
?microtable2.4 RTools
For Windows system, RTools (https://cran.r-project.org/bin/windows/Rtools/) is necessary to install some R packages from source (those that need compilation of C/C++ or Fortran code).
2.5 Dependence
2.5.1 Description
To keep the start and use of microeco package simplified, the installation of microeco only depend on several packages, which are compulsory-installed from CRAN and frequently used in the data analysis. So the question is that the user may encounter an error when using a class or function that invoke an additional package like this:
library(microeco)
data(dataset)
test <- trans_network$new(dataset = dataset, filter_thres = 0.001)
test$cal_network(network_method = "SpiecEasi")Error in test$cal_network(network_method = "SpiecEasi"): SpiecEasi package is not installed!
The reason is that network construction with ‘SpiecEasi’ method requires SpiecEasi package to be installed.
This package is deposited in GitHub and can not be installed automatically.
In addition, we donot put some packages released in CRAN and Bioconductor on the “Imports” part of microeco package.
The solutions:
Install the missing package when encounter such an error. Actually, it’s very easy to install the packages from CRAN or Bioconductor or Github. Just have a try.
Install all the packages in advance. This is recommended if the user is interested in most of the methods and want to run a large number of examples in this tutorial. If so, please read all of the following sections and install these packages.
2.5.2 CRAN packages
Some packages released in CRAN can not be installed automatically. These packages are necessary to reproduce some parts of the tutorial. If you want to install all of these packages or some of them, please run this:
# allow more waiting time to download each package
options(timeout = 1000)
# If a package is not installed, it will be installed from CRAN
# First select the packages of interest
tmp <- c("microeco", "mecoturn", "MASS", "GUniFrac", "ggpubr", "randomForest", "ggdendro", "ggrepel", "agricolae", "igraph", "picante", "pheatmap", "rgexf",
"ggalluvial", "ggh4x", "rcompanion", "FSA", "gridExtra", "aplot", "NST", "GGally", "ggraph", "networkD3", "poweRlaw", "ggtern", "SRS", "performance")
# Now check or install
for(x in tmp){
if(!require(x, character.only = TRUE)) {
install.packages(x, dependencies = TRUE)
}
}2.5.3 Bioconductor packages
Some dependent packages are deposited in bioconductor (https://bioconductor.org). Please run the following commands to install them one by one. Several packages may be installed from source. So, for the Windows system, please make sure RTools has been installed (https://chiliubio.github.io/microeco_tutorial/intro.html#rtools).
install.packages("BiocManager")
install.packages("file2meco", repos = BiocManager::repositories())
install.packages("MicrobiomeStat", repos = BiocManager::repositories())
install.packages("WGCNA", repos = BiocManager::repositories())
BiocManager::install("ggtree")
BiocManager::install("metagenomeSeq")
BiocManager::install("ALDEx2")
BiocManager::install("ANCOMBC")2.5.4 Github packages
A part of dependent packages in some methods comes from Github (https://github.com/). Each package from the GitHub platform is accompanied by installation instructions. However, due to the network instability of the platform, certain packages may fail to install online. As a result, in order to facilitate quick and convenient installation, we have collected these GitHub-dependent packages and consolidated them within a dedicated project repository (https://github.com/ChiLiubio/microeco_dependence). Please run the following commands to install them. For the Windows system, first make sure RTools has been installed (https://chiliubio.github.io/microeco_tutorial/intro.html#rtools).
# download link of the compressed packages archive
# Alternative from Gitee "https://gitee.com/chiliubio/microeco_dependence/releases/download/v0.20.0/microeco_dependence.zip"
url <- "https://github.com/ChiLiubio/microeco_dependence/releases/download/v0.20.0/microeco_dependence.zip"
# allow more time to download the zip file in R
options(timeout = 2000)
# Another way is to open the upper url in browser to download the zip file and move it to the current R working directory
download.file(url = url, destfile = "microeco_dependence.zip")
# uncompress the file in R
tmp <- "microeco_dependence"
unzip(paste0(tmp, ".zip"))
# install devtools
if(!require("devtools", character.only = TRUE)){install.packages("devtools", dependencies = TRUE)}
# run these one by one
devtools::install_local(paste0(tmp, "/", "SpiecEasi-master.zip"), dependencies = TRUE)
devtools::install_local(paste0(tmp, "/", "mixedCCA-master.zip"), dependencies = TRUE)
devtools::install_local(paste0(tmp, "/", "SPRING-master.zip"), dependencies = TRUE)
devtools::install_local(paste0(tmp, "/", "NetCoMi-main.zip"), repos = BiocManager::repositories())
devtools::install_local(paste0(tmp, "/", "beem-static-master.zip"), dependencies = TRUE)
devtools::install_local(paste0(tmp, "/", "chorddiag-master.zip"), dependencies = TRUE)
devtools::install_local(paste0(tmp, "/", "ggradar-master.zip"), dependencies = TRUE)
devtools::install_local(paste0(tmp, "/", "ggnested-main.zip"), dependencies = TRUE)
devtools::install_local(paste0(tmp, "/", "ggcor-1-master.zip"), dependencies = TRUE)2.5.5 Gephi
Gephi is an excellent network visualization tool and used to open the saved network file, i.e. network.gexf in the tutorial. You can download Gephi and learn how to use it from https://gephi.org/users/download/
2.5.6 Tax4Fun2
Tax4Fun2 is an R package for the the prediction of functional profiles and functional gene redundancies of prokaryotic communities (Wemheuer et al. 2020). It has higher accuracies than PICRUSt and Tax4Fun. The Tax4Fun2 approach implemented in microeco is a little different from the original package. Using Tax4Fun2 approach require the representative fasta file. The user do not need to install Tax4Fun2 R package again. The only thing need to do is to download the blast tool (ignore this if the blast tool has been in the path) and Ref99NR/Ref100NR database (select one). Download blast tools from “https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+” ; e.g. ncbi-blast-****-x64-win64.tar.gz for windows system. Note that some errors may come from the latest versions because of memory issue (https://www.biostars.org/p/413294/). An easy solution is to use previous version (such as 2.5.0). Download Ref99NR.zip from “https://cloudstor.aarnet.edu.au/plus/s/DkoZIyZpMNbrzSw/download” or Ref100NR.zip from “https://cloudstor.aarnet.edu.au/plus/s/jIByczak9ZAFUB4/download”. The alternative of the download link is “https://github.com/ChiLiubio/microeco_extra_data/releases/download/v1.0.0/Tax4Fun2_ReferenceData_v2.zip” or “https://gitee.com/chiliubio/microeco_extra_data/releases/download/v1.0.0/Tax4Fun2_ReferenceData_v2.zip”. Uncompress all the folders. The final folders should be like these structures:
blast tools:
|– ncbi-blast-2.5.0+
|—- bin
|—— blastn.exe
|—— makeblastdb.exe
|—— ……
Ref99NR:
|– Tax4Fun2_ReferenceData_v2
|—- Ref99NR
|—— otu000001.tbl.gz
|—— ……
|—— Ref99NR.fasta
|—— Ref99NR.tre
The path “Tax4Fun2_ReferenceData_v2” will be required in the trans_func$cal_tax4fun2() function. The blast tool path “ncbi-blast-2.5.0+/bin” is also required if it is not added to the system env path (environmental variable).
# Either seqinr or Biostrings package should be installed for reading and writing fasta file
install.packages("seqinr", dependencies = TRUE)
# or install Biostrings from bioconductor https://bioconductor.org/packages/release/bioc/html/Biostrings.html
# Now we show how to read the fasta file
# see https://github.com/ChiLiubio/file2meco to install file2meco
rep_fasta_path <- system.file("extdata", "rep.fna", package="file2meco")
rep_fasta <- seqinr::read.fasta(rep_fasta_path)
# or use Biostrings package
rep_fasta <- Biostrings::readDNAStringSet(rep_fasta_path)
# try to create a microtable object with rep_fasta
data("otu_table_16S")
# In microtable class, all the taxa names should be necessarily included in rep_fasta
otu_table_16S <- otu_table_16S[rownames(otu_table_16S) %in% names(rep_fasta), ]
test <- microtable$new(otu_table = otu_table_16S, rep_fasta = rep_fasta)
test2.6 Plot
Most of the plots in the package rely on the ggplot2 package system. We provide some parameters to optimize the corresponding plot, but it may be far from enough. The user can also assign the output a name and use the ggplot2-style grammers to modify it. Each data table used for visualization is stored in the object and can be saved for the customized analysis. Of course, the user can also directly modify the class and reload them to use. Any contribution of a modified class is appreciated via Github-Pull requests (https://github.com/ChiLiubio/microeco_tutorial/pulls) or Email (liuchi0426@126.com).
2.7 Rstudio
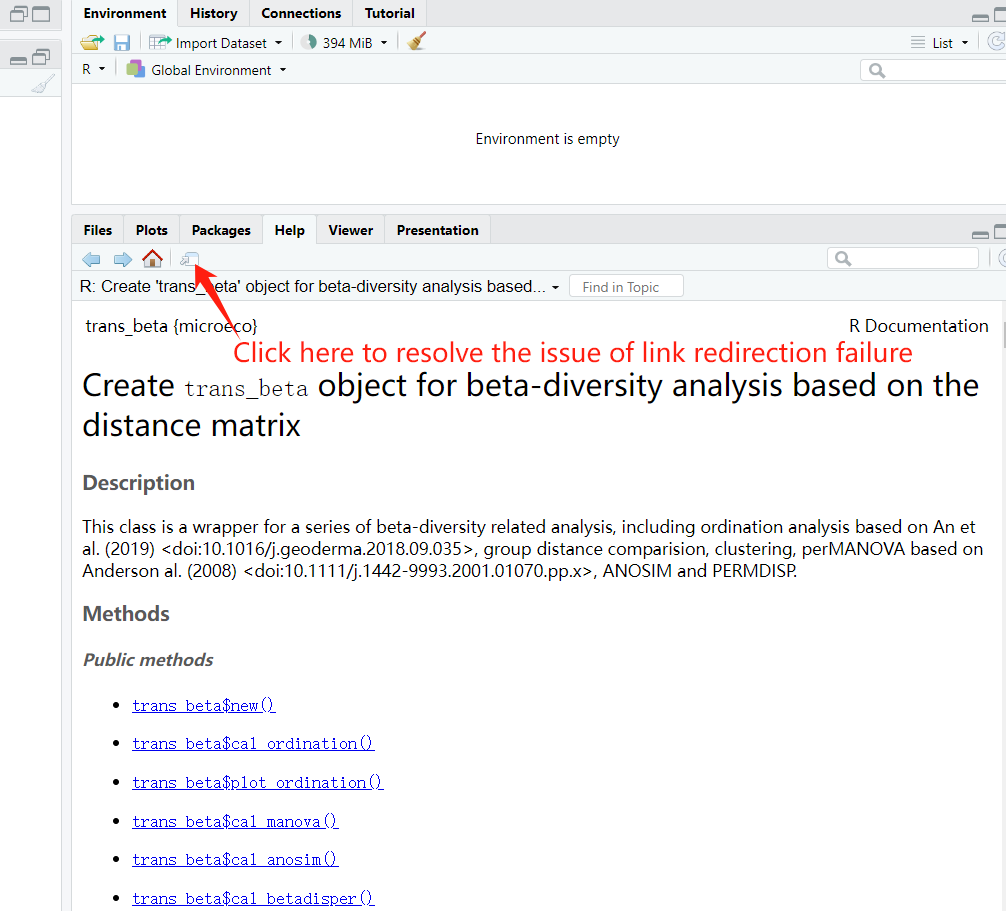
The modular design of help documentation can facilitate the document viewing. However, in Rtudio, there may be instances where the links fail to navigate properly. In such cases, the issue can be resolved by reopening a document window, as illustrated in the figure below.
2.8 Citation
To get the citation in R, please run:
citation("microeco")Please download the EndNote or other citation format from the publication website (https://doi.org/10.1093/femsec/fiaa255).